| Головна |
| «« | ЗМІСТ | »» |
|---|
Хвороба, названа метаболічним синдромом X, вперше описана в 1988 р як стан, що підвищує ризик розвитку цукрового діабету 2-го типу. Відмітною ознакою діабету 2-го типу є гальмування надходження глюкози в клітини через зниження чутливості тканин до інсуліну - інсулінорезистентності клітинних мембран. При цьому підвищується рівень інсуліну в крові (компенсаторна гіперінсулінемія). Інсулінорезі- стентності і компенсаторна гіперінсулінемія призводять до формування взаємопов'язаних порушень вуглеводного і жирового обміну, а також механізмів регуляції артеріального тиску.
Клінічними симптомами метаболічного синдрому X є гіперінсулінемія, порушення толерантності до вуглеводів, ожиріння, артеріальна гіпертонія, гіперурикемія і порушення системи фібринолізу, а також розвиток і м мунодефі цита.
Давно відома взаємозв'язок між ожирінням у чоловіків і серцево-судинними факторами ризику (гіперглікемією, гіперліпідемією і порушенням обміну сечової кислоти), а також більш високі концентрації інсуліну у хворих на артеріальну гіпертонію в порівнянні з нормотоніків.
У 50% хворих на цукровий діабет 2-го типу відзначається підвищення артеріального тиску, а захворюваність як артеріальною гіпертонією, так і на цукровий діабет зростає при збільшенні маси тіла. Беручи до уваги часте поєднання порушеної толерантності до глюкози, гіперінсулінемії, діслі- підем і артеріальної гіпертонії, зазначені стану об'єднали в поняття метаболічного синдрому X. Було встановлено, що основою розвитку цього комплексу симптомів є порушення чутливості тканин до інсуліну - ин- сулінорезістентность. Пізніше до метаболічного синдрому X були також віднесені зміни системи фібринолізу і гіперурикемія (рис. 13.12).
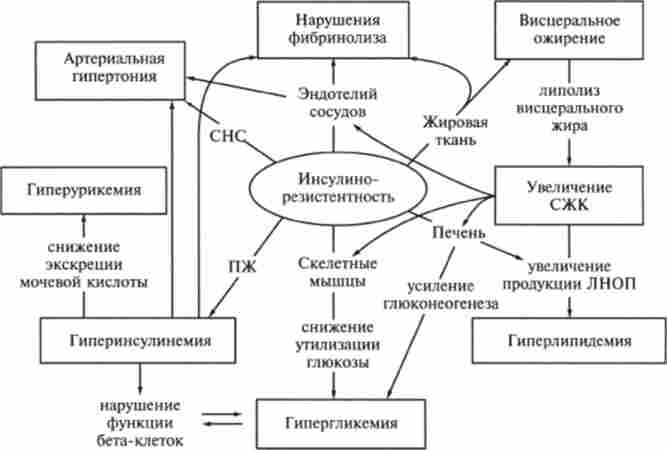
Мал. 13.12. Основні механізми розвитку метаболічного синлроме X.
ПЖ - підшлункова залоза, СНС - сімнашчсская нервова сіасма. СЖК - вільні жирні кіежпи, ЛНОП - ліпопротеїди дуже низької щільності
У розвитку метаболічного синдрому X мають значення як генетична схильність, так і особливості способу життя.
Для пояснення виникнення синдрому X висунута гіпотеза «економного» генотипу. Відповідно до цієї гіпотези в умовах дефіциту їжі інсуліно- резистентність забезпечує переваги для виживання. Вона сприяє збереженню нормогликемии і більш економного витрачання енергії в періоди голодування за рахунок зниження швидкості утилізації глюкози м'язовою тканиною, посилення глюконеогенезу і липогенеза.
У сучасному суспільстві в умовах харчового достатку, що сполучається з малорухливим способом життя, інсулінорезистентність втратила своє пристосувальне значення і призводить до розвитку ожиріння, цукрового діабету 2-го типу та атеросклерозу.
Одна з головних біологічних функцій інсуліну полягає в посиленні транспорту глюкози всередину клітини. У зв'язку з цим при інсулінорезистентності в першу чергу з'являються більш-менш виражені ознаки порушення вуглеводного обміну. В результаті зниження чутливості клітин-мішеней до дії інсуліну порушується засвоєння глюкози інсулінозалежний тканинами (печінкою, м'язами і жировою тканиною) і створюються передумови до розвитку гіперглікемії - підвищеного рівня глюкози в крові. Однак завдяки компенсаторного збільшення секреції інсуліну бета-клітинами підшлункової залози концентрація глюкози в сироватці крові протягом тривалого часу може залишатися нормальною. Таким чином, компенсаторна гіперінсулінемія є найбільш раннім і постійним симптомом інсулінорезистентності.
Протягом тривалого часу, а іноді довічно компенсаторна гіперінсулінемія сприяє збереженню нормогликемии і запобігає розвитку порушення толерантності до глюкози і цукрового діабету 2-го типу. Однак при наростанні інсулінорезистентності секреція інсуліну бета-клітинами виявляється недостатньою для подолання резистентності до гормону і підтримки нормоглікемії, т. Е. Розвивається відносний дефіцит інсуліну.
Нездатність бета-клітин підшлункової залози забезпечувати необхідний рівень гиперсекреции інсуліну служить основою для переходу від інсулінорезистентності до гіперглікемії (спочатку натщесерце, потім після харчової навантаження) і, нарешті, до явного цукрового діабету 2-го типу. При наявності порівнянної інсулінорезистентності у хворих на цукровий діабет 2-го типу здатність бета-клітин підтримувати компенсаторну гіперінсулінемію нижче, ніж у людей з порушенням толерантності до глюкози, і знижується в міру прогресування захворювання.
Найбільш ранні зміни функції бета-клітин, що спостерігаються у хворих з порушенням толерантності до глюкози, зачіпають в основному першу фазу секреції інсуліну - фазу швидкого вивільнення. Ці порушення можуть бути наслідком десенситизации бета-клітин підшлункової залози - оборотної їх рефрактерності до глюкози, яка розвивається при повторних епізодах гіперглікемії і характеризується зниженням здатності швидко реагувати на підвищення концентрації глюкози в крові.
У міру зростання порушень вуглеводного обміну і розвитку цукрового діабету з'являються незворотні зміни острівковогоапарату підшлункової залози, що супроводжуються збільшенням порушень інсуліносекреторной функції (ефект глюкозотоксичности).
Таким чином, виникає «порочне коло», що складається з підвищення рівня глюкози крові і зниження функції бета-клітин.
Іншим ключовим елементом патогенезу метаболічного синдрому X є вісцеральні ожиріння, що характеризується надмірним нагромадженням жирової тканини в черевній порожнині. Розподіл жирової тканини можна оцінити на підставі відносини об'єму талії до об'єму стегон. Вимірювання проводять за допомогою сантиметрової стрічки, що накладається циркулярно під краєм реберної дуги і на рівні кульшових суглобів відповідно. При наявності переважного накопичення жирової тканини в абдомінальній області цей показник перевищує 1,0 у чоловіків і 0,8 у жінок.
Розвиток вісцерального ожиріння пов'язують зі зміною метаболізму глюкокортикостероїдів в жировій тканині при інсулінорезистентності. Посилюється перетворення кортизону в кортизол під дією р-гідроксістероідредуктази-1, активність якої стимулюється інсуліном.
Внаслідок одночасного підвищення екскреції кортизолу з сечею його рівень в плазмі крові не підвищується, а гормон надає тільки місцеву дію на жирову тканину, стимулюючи диференціювання стромальних клітин в адіпоці- ти і внутрішньоклітинний накопичення ліпідів. Крім того, стимулюється перерозподіл жирової тканини по андроидное (чоловічому) типу з переважним накопиченням жирової клітковини в області верхньої половини тулуба і в черевній порожнині (в жировій тканині сальника і брижі).
Вісцеральні ожиріння вносить істотний внесок в розвиток і поглиблення інсулінорезистентності.
Чутливість p-адреноблокатори до катехоламінів і активність липо- протеідліпази в вісцеральної жирової тканини вище, ніж в підшкірному жировому шарі, тому процеси ліполізу тут протікають швидше.
Утворені в результаті ліполізу вільні жирні кислоти у великій кількості надходять по ворітної вени в печінку, а потім в системний кровотік.
Підвищення рівня вільних жирних кислот в портальному і системному кровотоці викликає ряд порушень вуглеводного і жирового обміну, а також зміни системи фібринолізу та функції ендотелію (рис. 13.12).
У печінки вільні жирні кислоти активізують глюконеогенез, що призводить до збільшення продукції глюкози печінкою і розвитку гіперглікемії натще. В кровотоці вони знижують активність фосфатидилинозитол-3-кінази інсулінового рецептора і пригнічують транспорт глюкози всередину клітин, що також сприяє розвитку гіперглікемії (ефект ліпотоксічності).
Показано, що при збільшенні відносини окружності талії і стегон, побічно характеризує кількість жирової тканини в області живота, чутливість скелетних м'язів до інсуліну і зв'язування інсуліну з рецепторами прогресивно погіршуються. Таким чином, збільшення цього індексу є клінічною ознакою інсулінорезистентності.
Підвищене надходження вільних жирних кислот в печінку і інсуліноре- резистентності гепатоцитів призводять до збільшення синтезу тригліцеридів (ТГ) і продукції печінкою ліпопротеїдів дуже низької щільності (ЛОНП). Порушується також кліренс ЛОНП і ліпопротеїдів низької щільності (ЛПНЩ) внаслідок зниження активності ліпопротеідліпази, яка контролюється інсуліном.
Підвищення секреції і порушення кліренсу ЛОНП є первинною ланкою в ланцюзі порушень обміну ліпопротеїдів при інсулінорезистентності. Зокрема, порушення кліренсу ЛОНП призводить до зниження рівня ліпопротеїдів високої щільності (ЛВП), так як вони утворюються в результаті переходу апопро- Теїн і фосфоліпідів з ЛОНП і ЛНП в процесі їх ліполізу.
Вважається також, що підвищення рівня ЛОНП є основною причиною появи структурних змін ЛНП у хворих з інсулінорезистентністю. В результаті обміну між ефірами холестерину, ЛНЩ і трігліцерідамі ЛОНП відбувається насичення ЛНП трігліцерідамі, а потім, в результаті ліполізу, вміст тригліцеридів в складі ЛНП знижується, що призводить до формування більш дрібних і щільних ЛНП з високим вмістом білка арів 100 і низьким вмістом ефірів холестерину .
Таким чином, основними характеристиками дисліпідемії при синдромі інсулінорезистентності є гіпертригліцеридемія, підвищення рівня ЛОНП і ЛНП, зниження рівня ЛВП і зміна структури ЛНП.
Метаболічні порушення при інсулінорезистентності не обмежуються змінами вуглеводного і жирового обміну і зачіпають також систему фіб- ріноліза і механізми регуляції артеріального тиску.
Для інсулінорезистентності характерно зниження фібринолітичної активності крові. Це пов'язано зі збільшенням під дією інсуліну рівня проінсуліну, а також фактора а некрозу пухлини і трансформуючого фактора р зростання. Ці фактори в великій кількості синтезуються в жировій тканині при ожирінні. Зростає продукція інгібітора тканинного активатора плазміногену 1-го типу в ендотелії, гепатоцитах і жирової тканини. Розвиток артеріальної гіпертонії при інсулінорезистентності може бути результатом порушення рівноваги між прогіпертензівним і гіпотензивний ефекти інсуліну.
Інсулінорезистентність характеризується тканинної специфічністю і зачіпає тільки певні органи і клітини-мішені (скелетні м'язи, ендотелій і гладком'язові клітини судин, жирова тканина, печінка). В інших органах і системах стимульовані інсуліном процеси можуть навіть активуватися внаслідок компенсаторної гіперінсулінемії.
Гиперинсулинемия сприяє підвищенню артеріального тиску за рахунок збільшення реабсорбції натрію і води нирками, стимуляції центрів симпатичної нервової системи і активації обміну іонів натрію і водню в клітинах гладеньких м'язів судин. Це призводить до накопичення в клітинах іонів натрію і кальцію і підвищення чутливості до пресорну дію катехоламінів і ангіотензину II. Крім того, інсулін, який діє на локальну рснінангіотен- Зіновій систему судин, стимулює зростання і проліферацію клітин гладких м'язів і сприяє розвитку гіпертрофії медії і звуження внутрішнього діаметра судин, що призводить до постійно підвищеного артеріального тиску.
Вплив інсуліну на артеріальний тиск неоднозначно. У ряді клінічних і експериментальних досліджень було показано, що інсулін може давати значимі вазодилатаційний і гіпотензивний ефекти. Вважають, що інсулін викликає релаксацію судин шляхом прямого впливу на клітини гладеньких м'язів судин або вивільнення медіаторів з ендотелію.
Крім того, вважається, що інсулін є стимулятором синтезу і секреції ендотеліального фактора релаксації - оксиду азоту (N0), який грає важливу роль в регуляції системної і ниркової гемодинаміки. При інсулінорезистентності продукція NO ендотелієм знижується, в результаті чого порушуються процеси ендотелійзалежної вазодилатації. Дана обставина може бути пов'язано як зі зменшенням стимулюючого впливу інсуліну на синтез і секрецію NO внаслідок інсулінорезистентності судинної стінки, так і з гнітючим впливом вільних жирних кислот на активність МО-синтетази.
Інсулін викликає також вивільнення з ендотелію ендотеліну-1, що володіє вазоконстрикторні властивості. При інсулінорезистентності може порушитися рівновага між индуцируемой інсуліном секрецією N0 і ендотеліну-1 із зсувом в сторону останнього. NO крім вазодідатірующего дії володіє також антиатерогенні властивостями, а ендотелії-1, навпаки, дає вазоконстрикторний і мітогенний ефекти, тому порушення цієї рівноваги може сприяти розвитку не тільки артеріальної гіпертонії, а й атеросклерозу.
Перераховані вище зміни вуглеводного, жирового обміну і функції ендотелію, які виявляються у хворих з метаболічним синдромом і цукровим діабетом 2-го типу, мають важливе клінічне значення, так як призводять до формування цілого комплексу чинників ризику, що створюють передумови для розвитку атеросклерозу, артеріальної гіпертонії і їх серцево-судинних ускладнень.
У хворих на цукровий діабет 2-го типу ризик розвитку ішемічної хвороби серця (ІХС) підвищено в 3 рази, а у хворих з порушенням толерантності до глюкози - в 2 рази. Однак навіть при відсутності порушення толерантності до глюкози, коли єдиною ознакою труднощі утилізації глюкози тканинами є компенсаторна гіперінсулінемія, ризик розвитку серцево-судинних ускладнень вже значно підвищено. У цей період формується комплекс факторів ризику атеросклерозу, обумовлених інсулінорезистентністю: гіпертонія. гіперурикемія, порушення ліпідного спектра та системи фібринолізу.
Порушення толерантності до глюкози супроводжується різким збільшенням частоти макросудинних ускладнень, а до моменту розвитку хронічної гіперглікемії, що дозволяє діагностувати цукровий діабет, у багатьох пацієнтів вже є ІХС, в тому числі інфаркт міокарда в анамнезі. Дана обставина підкреслює необхідність своєчасної діагностики метаболічного синдрому X та корекції пов'язаних з ним метаболічних порушень.
Питання і завдання до paid. 13.4