До зазначених захворювань належать моногенно зумовлені патологічні стани, успадковані відповідно до законів Менделя.
Залежно від функціональної значущості первинних продуктів відповідних генів генні хвороби поділяють на спадкові порушення ферментних систем (ензимопатії), дефекти білків крові (гемоглобінопатії), дефекти структурних білків (колагенові хвороби) і генні хвороби з нез'ясованим первинним біохімічним дефектом.
В основі ензимопатія лежать або зміни активності ферменту, або зниження інтенсивності його синтезу. У гетерозигот - носіїв мутантного гена присутність нормального алеля забезпечує збереження близько 50% активності ферменту у порівнянні з нормальним станом. Тому спадкові дефекти ферментів клінічно проявляються у гомозигот, а у гетерозигот недостатня активність ферменту виявляється спеціальними дослідженнями.
Залежно від характеру порушення обміну речовин в клітинах серед ензимопатія розрізняють наступні форми.
1. Спадкові дефекти обміну вуглеводів (галактоземія - порушення метаболізму молочного цукру - лактози; мукополіса- харідози - порушення розщеплення полісахаридів).
2. Спадкові дефекти обміну ліпідів і ліпопротеїнів (сфінголіпідози - порушення розщеплення структурних ліпідів; порушення обміну ліпідів плазми крові, що супроводжуються збільшенням або зниженням в крові холестерину, лецитину).
3. Спадкові дефекти обміну амінокислот (фенілкетону- рія - порушення обміну фенілаланіну (див. Розд. 4.1); тирозиноз - порушення обміну тирозину; альбінізм - порушення синтезу пігменту меланіну з тирозину і ін.).
4. Спадкові дефекти обміну вітамінів (гомоцістіну- рія - розвивається як результат генетичного дефекту коферменту вітамінів В6 і В12, успадковується по аутосомно-рецесивним типом).
5. Спадкові дефекти обміну пуринових і піримідинових азотистих основ (синдром Леша - Найя, пов'язаний з недостатністю ферменту, який каталізує перетворення вільних пуринових підстав в нуклеотиди, успадковується по Х-зчепленим рецесивним типом).
6. Спадкові дефекти біосинтезу гормонів (адреногені- тальний синдром, пов'язаний з мутаціями генів, які контролюють синтез андрогенів; тестикулярная фемінізація, при якій не утворюються рецептори андрогенів).
7. Спадкові дефекти ферментів еритроцитів (деякі гемолитичні несфероцітарние анемії, які характеризуються нормальною структурою гемоглобіну, але порушенням ферментної системи, яка бере участь в анаеробних (безкисневих) розщепленні глюкози. Успадковуються як по аутосомно-рецесивним, так і по Х-зчепленим рецесивним типом).
Гемоглобинопатии - це група спадкових захворювань, що викликаються первинним дефектом пептидних ланцюгів гемоглобіну і пов'язаним з цим порушенням його властивостей і функцій. До них відносять метгемоглобінемії, еритроцитоз, серповидно-клітинну анемію, таласемії (див. Розд. 4.1).
В основі виникнення колагенових захворювань лежать генетичні дефекти біосинтезу і розпаду колагену - найважливішого структурного компонента сполучної тканини. До цієї групи відносять хвороба Еллерс - Данлоса, що характеризується великим генетичного поліморфізму і спадщини як по аутосомно-домінантним, так і по аутосомно-рецесивним типом, хвороба Марфана, успадковується за аутосомно-домінантним типом, і ряд інших захворювань.
Спадкові хвороби з нез'ясованим первинним біохімічним дефектом. До цієї групи належить переважна більшість моногенних спадкових хвороб. Найбільш поширеними є наступні.
1. муковісцидоз - зустрічаються з частотою 1: 2500 новонароджених; успадковуються по аутосомно-рецесивним типом. В основі патогенезу захворювання - спадкове ураження екзокринних залоз і залізистих клітин організму, виділення ними густого, зміненого за складом секрету і пов'язані з цим наслідки.
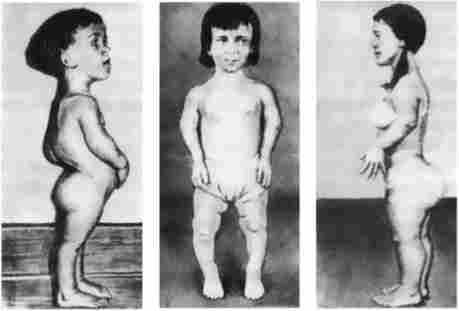
2. Ахондроплазія - захворювання, в 80-95% випадків обумовлене знову виникла мутацією; успадковується по аутосомно-домінантним типом; зустрічається з частотою приблизно 1: 100 000. Це захворювання кісткової системи, при якому спостерігаються аномалії розвитку хрящової тканини переважно в епіфізах трубчастих кісток і кістках основи черепа (рис. 7.23).
3. м'язові дистрофії (міопатії) - захворювання, пов'язані з ураженням поперечно-смугастих і гладких м'язів. Різні форми характеризуються різним типом успадкування. Наприклад, прогресуюча псевдогіпертрофічна міопатія Дюшена успадковується по Х-зчепленим рецесивним типом і проявляється переважно у хлопчиків на початку першого десятиліття життя.
Мал. 7.23. Хворі з ахондроплазією
Відома м'язова псевдогіпертрофічна дистрофія, успадковується за аутосомно-рецесивним типом, яка починає розвиватися в другій половині першого десятиліття життя і зустрічається з однаковою частотою в обох статей. М'язова дистрофія плечового і тазового пояса: успадковується по аутосомно-домінантним типом ит. д.
Вивчення спадкових захворювань у людини свідчить про те, що нерідко подібне фенотипічніпрояв хвороби буває обумовлено декількома різними мутаціями. Це явище вперше було описано в 30-х рр. С. Н. Давиденкова і названо генетичною гетерогенністю спадкових захворювань. Генетична гетерогенність спадкових хвороб може бути обумовлена мутаціями різних генів, що кодують ферменти одного метаболічного шляху, а також мутаціями одного і того ж гена, що приводять до появи різних його алелей.
Серед розглянутих вище спадкових хвороб особливо високим ступенем генетичного поліморфізму відрізняються муко- полісахарідози, генетична різнорідність яких пояснюється множинними мутаціями в 11-12 генах, пов'язаних загальною функцією розщеплення полісахаридів. Великий генетичною гетерогенністю характеризується вроджена аутосомно-рецесивна форма глухоти, при якій розрізняють не менше 35 генетично різних варіантів з фенотипично подібним проявом.
Великі перспективи в розшифровці спадкової гетерогенності генних хвороб відкриваються в зв'язку з застосуванням молекулярно-генетичних методів їх прямого аналізу за допомогою ДНК-зондів.
Різноманітність клініки спадкових хвороб проявляється у відмінності часу початку захворювання, в спектрі і ступеня вираженості симптомів, в перебіг та наслідки у різних хворих. Наприклад, що успадковується по аутосомно-домінантним типом хорея Гентингтона, при якій дивуються базальні ганглії головного мозку, клінічно починає проявлятися у вигляді мимовільних рухів в різному віці, але частіше в 40-45 років. З часом почала клінічного прояву пов'язана і тяжкість перебігу захворювання (див. 7.4.1.4).
Про клінічному поліморфізм можна говорити лише відносно генетично визначеною спадкової форми. Причини клінічного поліморфізму можуть бути як генетичними, так і середовищні. До генетичних причин можна віднести дію генів-модифікаторів на прояв патологічно зміненого гена і складну систему різноманітних взаємодій між ним та іншими генами. Крім того, різноманітність клінічного прояву спадкових захворювань може залежати від факторів середовища, в якій розвивається організм і яка впливає на прояв патологічно змінених генів.
Головний мозок. Проміжний мозок - анатомія центральної нервової системи В результаті вивчення даного розділу студент повинен: знати основні області проміжного мозку, їх взаємне розташування і зв'язку з іншими відділами головного мозку; загальну анатомію, афференти і ефферентов таламуса, його основні ядра (на анатомічному рівні і в зв'язку з виконуваними функціями
Головний мозок. Кінцевий мозок (великі півкулі) - анатомія центральної нервової системи В результаті вивчення даного розділу студент повинен: знати загальні анатомічні особливості кінцевого мозку; різноманітність сірого (кора і базальні ядра) і білої речовини; будову і функції базальних ядер; що входять до їх складу структури і їх зв'язку; сучасні уявлення про палео,
Глюкагон - біохімія Глюкагон синтезується в а-клітинах острівців підшлункової залози. Це пептидний гормон, що складається з 29 амінокислотних залишків з молекулярною масою 3,5 kDa. Нижче наведена амінокислотна послідовність глюкагону людини: Біосинтез. Глкжагон, подібно до багатьох біологічно активних пептидів,
Глікопротеїни мембран еукаріотичної клітини - біохімія людини Клітини тварин оточені м'якою, гнучкою структурою - клітинної мембраною (див. Рис. 6.2). Вона складається з речовин, повністю відрізняються за складом від мембран у твердій клітинної стінки рослин і мікроорганізмів. У клітинній оболонці еукаріотів вуглеводи пов'язані з мембранними білками,
Гліальні клітини - нервова система: анатомія, фізіологія, Нейрофармакологія До клітинам нервової тканини крім нейронів відносяться також гліальні клітини, які дуже різноманітні за будовою і виконуваних функцій. гліальні клітини - нейроглії, гліоціти - мають багато спільних рис з нейронами, зокрема для них характерна наявність великої кількості розгалужених відростків
Гіпоталамус і середній мозок - нейрофізіологія У гл. 11 було показано, що в гіпоталамусі виділяють кілька центрів, пов'язаних з тими чи іншими емоційними реакціями. Так, у кішок роздратування гіпоталамуса може супроводжуватися агресивною поведінкою з зовнішніми ознаками люті або оборонним поведінкою із зовнішніми ознаками страху. Якщо
Гіпофіз, гіпоталамо-гіпофізарна система - нейрофізіологія Розташований гіпофіз в так звані турецькому сідлі основної кістки черепа і як би висить на ніжці під гіпоталамусом. У ньому розрізняють передню частку ( аденогипофиз ) І задню частку ( нейрогипофиз ). Вони розвиваються з різних ембріональних зачатків. Задня частка гіпофіза в процесі еволюції
Геномний рівень організації спадкового матеріалу, геном. Генотип. Каріотип - біологія. Частина 1 геномом називають всю сукупність спадкового матеріалу, укладеного в гаплоидном наборі хромосом клітин даного виду організмів. Геном видоспецифичен, тому що являє собою той необхідний набір генів, який забезпечує формування видових характеристик організмів в ході їх нормального онтогенезу