| Головна |
| «« | ЗМІСТ | »» |
|---|
Завдяки великому обсягу людського генома і більш низькому тиску природного відбору, пов'язаного з соціальною природою людини (див. Розд. 1.9), в генофонді людства за тисячоліття його існування в результаті постійно йде мутаційного процесу накопичено велику кількість алелей багатьох генів. Це є причиною формування у людей різноманітних варіантів ознак і властивостей як на структурному, так і на біохімічному рівнях. В основі індивідуальних відмінностей за багатьма білків лежать зміни відповідних генів. Вивчення амінокислотного складу варіантів білків людського організму, інтенсивності їх синтезу, функціональної активності дає цінні відомості про організацію і експресії його спадкового матеріалу.
Зручною моделлю для вивчення молекулярно-генетичних механізмів спадковості і мінливості у людини є гемоглобін - специфічний білок еритроцитів, легко виділяється з організму без застосування трудомістких біохімічних методик. В результаті тривалого вивчення цього білка накопичилося багато фактів, які свідчать про мінливість його первинної структури і властивостей. В даний час виявлено близько 400 різних різновидів гемоглобіну, що зустрічаються як у нормальному розвитку на різних стадіях онтогенезу, так і призводять до різних захворювань.
Молекула гемоглобіну складається з чотирьох поліпептидних ланцюгів (двох а- і двох 3-ланцюгів), кожна з яких з'єднана з небілковим компонентом - гемом, що містить залізо. Дві названі поліпептидні ланцюги мають варіанти, контрольовані різними, але близькими нуклеотидними послідовностями, які утворюють два сімейства генів (див. Розд. 3.6.4.3). Різні нуклеотидні послідовності експресуються на певних стадіях індивідуального розвитку - у ембріона, плода, після народження (див. Розд. 6.2). При цьому поліпептиди, що змінюються в залежності від стадії онтогенезу, незначно відрізняються за амінокислотним складом. так, Ау- і ° у-Глобине розрізняються по одній амінокислоті в 136-му положенні (аланін або гліцин). Варіанта-глобіну
(ТАу) в 75-му положенні замість изолейцина має треонин. Ланцюг б відрізняється від (5-ланцюга лише десятьма амінокислотними залишками.
З численних мутацій гемоглобіну більшість досить рідкісні і лише деякі з них зустрічаються частіше інших, наприклад HbS, НЬС, нье. Велика частина варіантів гемоглобіну (близько 350) різниться одиничними амінокислотними замінами, причиною яких є генні мутації, пов'язані з заміною підстав в нуклеотидних послідовностях а- або Р-глобинового сімейства. Багато амінокислотні заміни суттєво не впливають на функцію гемоглобіну і не призводять до патологічних проявів. Як правило, це заміни в звернених назовні ділянках поліпептидних ланцюгів тетрамера.
Заміни амінокислот, що порушують нормальну спіральну структуру ланцюгів, часто викликають нестійкість гемоглобіну. Заміна в ділянках, якими а- і P-ланцюга контактують один з одним, впливають на спорідненість гемоглобіну до кисню. Порушення функцій гемоглобіну, що виникають в результаті таких змін структури а- і Р-глобінових генів, ведуть до появи захворювань, які можна розділити на чотири основні групи.
Захворювання перших трьох груп успадковуються за домінантним типом, так що гетерозиготи по мутантного гену страждають порушенням здоров'я. Спадкування серповидно-клітинної анемії при звичайних умовах здійснюється за рецесивним типом, але в умовах сильної гіпоксії, наприклад при знаходженні на висоті понад 3000 м над рівнем моря гетерозиготи HbA HbS також страждають на анемію.
Описані мутантні форми гемоглобіну виникають в результаті змін структури генів за типом заміни підстав. Мутації іншого характеру призводять до появи алелей глобинов, що обумовлюють інші види патології. Так, порушення процесу рекомбінації між аллельними генами (нерівноцінний кросинговер) призводить до зміни числа нуклеотидів в них. Наслідком цього може бути зрушення рамки зчитування. Нерідко результатом таких структурних змін генів є пригнічення синтезу тієї чи іншої ланцюга гемоглобіну, що приводить до розвитку патологічних станів, відомих під загальною назвою талассемии.
Делеция одного нуклеотиду в 139-м триплеті а-глобинового гена, що складається з 141 триплета, призводить до зсуву рамки зчитування і прочитування в новій рамці терминирующего 142-го кодону. При цьому а-глобінових ланцюг подовжується на п'ять додаткових амінокислот. Такою особливістю a-ланцюгів характеризується гемоглобін Vayne. Коли делеция розташовується ближче до 5'-кінця, активний продукт не синтезується і розвиваються різні форми а-, (3- і у-талассемий.
Деякі варіанти гемоглобинов виникають в результаті дуплікацій. Так, гемоглобін Grady несе дуплікацію 116-118 амінокислотних залишків в a-ланцюга. У гемоглобіні Cranston подовження p-ланцюга до 158 амінокислотних залишків є результатом дуплікації AG-послідовності після 144-го триплета і подальшого зсуву рамки з просочуванням термінального кодону.
Описане вище свідчить про те, що різні відхилення в структурі ДНК глобінових генів призводять до заміни амінокислот або подовженню поліпептидних ланцюгів. Це є причиною утворення багатьох варіантів гемоглобіну, які визначають розвиток у людини захворювань, наследующихся в ряду поколінь.
Не менший інтерес представляють механізми розвитку різних захворювань людини, в основі яких лежать мутації генів, що призводять до синтезу білків-ферментів зі зниженою активністю або до його придушення. Це порушує перебіг процесів, що каталізують даними ферментами в клітинах організму. Прикладом спадково детермінованих ушкоджень метаболізму в організмі людини служить фенілкетонурія, що розвивається внаслідок порушення процесів обміну амінокислоти фенілаланіну і накопичення в організмі токсичних проміжних продуктів.
При дефекті ферменту фенілаланінгідроксилази фенілаланін чи не перетворюється в тирозин (рис. 4.1) і накопичується в крові хворих в великих концентраціях (до 0,5-0,6 г / л замість 0,003- 0,04 г / л в нормі). Це призводить до часткового перетворення фенілаланіну в фенилуксусной і фенілмолочная кислоти, накопичення яких поряд з підвищеною концентрацією самого фенілаланіну токсично діє на мозок дитини. В результаті у дітей спостерігається різна ступінь дефекту розумового розвитку. Порушення метаболізму фенілаланіну супроводжується також порушенням синтезу пігменту меланіну, тому у хворих спостерігається слабка пігментація волосся і райдужної оболонки очей. Крім того, висока концентрація фенілаланіну надає інгібуючий вплив на ряд ферментних систем, що беруть участь у перетворенні інших амінокислот: у хворих розвивається судомний синдром, наростає відставання інтелектуального розвитку. Спадкування фенілкето- Нурии здійснюється за рецесивним типом.
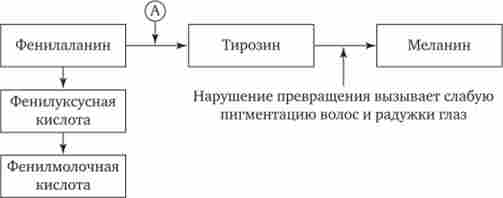
Мал. 4.1. Коротка схема обміну фенілаланіну.
А - фермент фенілаланінгідроксилази, спадковий дефект якого призводить до розвитку фенілкетонурії
Таким чином, розглянуті вище приклади демонструють весь спектр дії молекулярно-генетичних механізмів, що забезпечують освіту в людському організмі білків, як нормально функціонуючих, так і обумовлюють розвиток різних патологічних станів. Зі сказаного з приводу гемоглобіну слід, що, по-перше, освіта головного функціонального білка еритроцитів знаходиться під генним контролем, по-друге, формування тетрамер- ної форми цього білка, з якою пов'язана його фізіологічна активність, вимагає взаємодії неалельних генів а- і ( 3-глобинов.
Специфічний контроль небілкової частини молекули гемоглобіну також має місце і здійснюється незалежно, через гени ферментів, необхідних для синтезу гема. Особливості прояви патологічних ознак у носіїв мутантних алелів свідчать про існування певних відносин між ними і нормальними алелями. Так, аллель серповидно-клеточно- сти в поєднанні з нормальним алелем (3-глобіну (HbA HbS) проявляє себе в звичайних умовах як рецесивний. Так само поводиться мутантний аллель гена, детермінують синтез ферменту фені- лаланінгідроксілази. Проявом взаємодії між мутантним і нормальним алелями за типом домінування останнього є формування в організмі білка з нормальними властивостями у гетерозигот. Відсутність нормального алеля в генотипі організму, гомозиготного по мутантного аллели, призводить до розвитку патологіческог про стан, обумовленого порушенням функціональної активності відповідного білка.
Особливу групу спадково обумовлених патологічних станів у людини представляють захворювання, причиною яких є мутації мітохондріальної ДНК (мтДНК).
Біосинтез мітохондріальних білків знаходиться під контролем двох генетичних систем: ядерних і мітохондріальних генів.
Велика частина білків кодується ядерної ДНК, синтезується в цитоплазмі, а потім транспортується в мітохондрії. Поряд з цим в кільцевої молекулі ДНК органели є гени, які відповідають за власний синтез білків, а також беруть участь в ньому тРНК і рРНК. У ядерному геномі є значна кількість генів, які забезпечують функціонування мітохондріальної ДНК. Припускають, що мутації деяких ядерних генів призводять до ділок значних ділянок ДНК мітохондрій. В результаті порушується синтез власних білків, до числа яких відносяться і ферменти дихальних ланцюгів, порушується дихальна функція мітохондрій.
У людини описано понад 100 захворювань, причиною яких є зміни в структурі мтДНК (див. Розд. 6.4.1.4).