| Головна |
| «« | ЗМІСТ | »» |
|---|
деменцією називають зниження інтелекту, при якому порушуються пам'ять, логіка, мислення, здатності до навчання. Зазвичай деменція розвивається у людей похилого віку, і цей процес розвивається повільно.
Іноді деменція може виникати і у молодих людей під дією отруєння або важкої травми мозку. Звичайно, з віком пам'ять страждає у всіх людей, і не треба плутати цей природний процес з важким зниженням розумових здібностей, яким характеризується деменція.
Хвороба Альцгеймера. Найбільш часто зустрічається причина деменції - хвороба Альцгеймера (сенільний деменція). Зараз на це захворювання страждають більше 30 млн, головним чином, літніх людей. За різними відомостями від 3 до 10% людей старше 65 років в тій чи іншій мірі уражені цим нейродегенеративних недугою. Найчастіше хвороба Альцгеймера спостерігається у жінок, хоча, мабуть, це пов'язано з тим, що жінки живуть в середньому набагато довше за чоловіків, і хвороба встигає розвинутися. Іноді деменція по Альцгеймера типу розвивається і у людей, які не досягли 40 років, хоча це трапляється відносно рідко.
До типових симптомів хвороби Альцгеймера відносяться: погіршення уваги і пам'яті, дезорієнтація в часі і в просторі, погіршення мови, порушення координації рухів, розлади емоцій (депресії, невмотивована агресія). Поступово хворий стає інвалідом, які потребують постійного нагляду і догляду. Хвороба може розвиватися протягом кількох років.
При посмертному розтині в мозку видно численні сліди атрофії: розширення мозкових шлуночків, звуження звивин кори великих півкуль, руйнування гіпокампу.
На клітинному рівні маркерами хвороби Альцгеймера служать багатоклітинні бляшки з білка - [3-амілоїду, розташовані в міжклітинному просторі в паренхімі мозку (сенільні бляшки), а також внутрішньо-нейронні клубки з спірально скручених ниток фосфорилированного білка цитоскелету - тау (т). Найбільші порушення тканин мозку простежуються у фронтальних областях кори великих півкуль і гіпокампі.
У мембрані нейронів виявлено білок - попередник р-амілоїду, що включає в себе 695 амінокислот. Це білок, мабуть, виконує кілька функцій, зокрема, взаємодіючи з З ^ -белком, підвищує рівень циклічного гуанозинмонофосфату (цГМФ) в клітці і уповільнює надходження іонів кальцію. Таким чином, цей білок-попередник р-амілоїду (АРР, від англ. Amyloid Precursor Peptide) Виконує захисну функцію при збудження. АРР рано чи пізно піддається розпаду під дією ферментів-секретази. У здорових людей при цьому утворюються три розчинних фрагмента, які, можливо, беруть участь в утворенні міжклітинних контактів. У хворих людей процес розпаду (протеолізу) АРР йде з утворенням двох розчинних фрагментів і одного нерастворимого, що складається з 42 або 43 амінокислот (А | 342 (43)). У міру накопичення цього «неправильного» фрагмента він починає утворювати амілоїдні волокна, що створюють амілоїдні бляшки. Треба сказати, що Ар42 (43) накопичується в мозку всіх літніх людей, проте у здорових осіб його дуже мало, і він не має ніякого патогенної ролі. У регуляції розпаду АРР беруть участь Mj-холінорецептори. Активація холінергічної системи через протеинкиназу З підсилює фосфорилювання АРР, що стимулює його «правильний» розпад. При зниженні активності холінергічної системи спостерігаються порушення в процесі протеолізу і освіту А (342 (43).
Як же проявляється токсична дія Ар42 (43)? Підвищується активність системи збуджуючих амінокислот - аспартату і, головне, глутамату. Тривале відкриття каналів iVMDA-рецепторів призводить до неконтрольованого потоку кальцію в клітини і підвищення рівня внутрішньоклітинного кальцію до токсичного.
Підвищується рівень NO в клітинах, в результаті чого страждають зовнішні мембрани, як самої клітини, так і її мітохондрій. Це призводить до зменшення синтезу АТФ і зниження активності АТФ-залежних ферментів Na+, До+-АТФ-ази, що викликає повільну деполяризацію клітинних мембран, і це знову-таки підсилює потік іонів кальцію в клітину. Формуються атипові катіонні канали, через які потік Са2+ і Na+ ще більше деполяризує клітку. Вплив Ар42 (43) на обмін в нервових клітинах викликає посилене фосфорилювання білка т і його агрегацію в нейрофібрилярних клубки, які відкладаються в цитоплазмі нейронів, ще більше дезорганизуя їх роботу.
Всі ці порушення, хоч і розвиваються дуже повільно, руйнують мозок і призводять до смерті хворого.
В основі значної частини випадків захворювання хворобою Альцгеймера лежать мутації, що дісталися нам від предків. Близько половини хворих в літньому віці не мають родичів, що страждали на цю недугу. Але у 30-40% хворих явно простежується позитивний сімейний анамнез. Що ж стосується ранніх форм захворювання (до 58 років), то встановлено, що в цьому випадку хвороба має генетичну основу, і вже знайдені, як мінімум, три гена в 1, 14 і 21-й хромосомах, мутації яких призводять до раннього розвитку хвороби Альцгеймера. Мутація одного гена (21-я хромосома) тягне за собою порушення в первинній структурі АРР, а два гена кодують структуру регуляторних білків - пресеніліна 1 і пресеніліна 2. Ці білки, зокрема, регулюють процесинг і транспорт ряду мембранних білків, в тому числі і білка - попередника р-амілоїду - АРР. Тому їх мутації обумовлюють викривлення в структурі фрагментів АРР. Цікаво, що у хворих на синдром Дауна, в клітинах яких знаходяться три хромосоми 21-й пари, швидкість утворення АРР набагато вище, ніж в нормі, і в великих кількостях накопичуються продукти його розпаду, що тягне за собою раннє слабоумство.
Як же постаратися уникнути хвороби Альцгеймера, адже ніяких специфічних способів її лікування не існує? Треба пам'ятати, що посилена робота мозку покращує його кровопостачання, знижує ризик склерозу. Тому люди, постійно «навантажують» свій мозок, хворіють набагато рідше і пізніше, ніж «ледарі». Бажано уникати струсів мозку, так як всі види деменції у боксерів спостерігаються частіше. Хвороба провокується у тих людей, хто змушений постійно працювати з різними розчинниками, бензином, ацетоном, клеями і, звичайно, у токсикоманів. У всіх цих людей спостерігається хронічне ураження билипидного шару мембран клітин мозку і, як наслідок, недоумство. Специфічного захисту, як уже говорилося, немає. Однак деякі мікроелементи (цинк, магній), амінокислоти (метіонін) і вітаміни (С, Е) зменшують ймовірність виникнення хвороби Альцгеймера, відсувають початок хвороби і гальмують її розвиток.
Пріонні хвороби. Смертельними пейродегенератівнимі захворюваннями є так звані губчасті енцефалопатії, які характеризуються руйнуванням нервової тканини, в результаті чого мозок набуває губчасту консистенцію, в різних структурах накопичуються стійкі до розпаду фібрили, рідше - бляшки. У цих бляшках немає (3-амілоїду. На відміну від хвороби Альцгеймера губчасті енцефалопатії зустрічаються у людини дуже рідко і можуть передаватися інфекційним шляхом. Дослідження причин цих захворювань показано, що вони виникають або через спонтанної модифікації звичайного клітинного білка - приона - в патогенну форму, або через зараження таким «неправильним» пріонних білком вже зі зміненою конформацией молекули.
У людей до таких хвороб відносяться:
Подібні захворювання описані і у деяких тварин, зокрема губчаста хвороба мозку корів ( «коров'ячий сказ»). Першою з цих хвороб була описана і досліджена куру.
Історія досліджень куру могла б лягти в основу прекрасного детективного роману.
У 1956 р австралійські лікарі звернули увагу на дивну хворобу, яка вражала тільки аборигенів з племені форі, що населяє віддалені і дуже важкодоступні гірські райони північного сходу Нової Гвінеї. Місцеві жителі називали цю хворобу «куру», що означає «смерть, що сміється». Хворі люди невпевнено рухалися, худнули, втрачали інтерес до життя, у них погіршувалася пам'ять. Але найхарактернішим симптомом був безпричинний сміх, який час від часу стрясав хворого. Забігаючи вперед, слід сказати, що через кілька років з'ясувалося, що цей сміх був судоми м'язів обличчя і не мав нічого спільного з істинним веселощами. Хвороба тривала, постійно посилюючись, рік або більше, а потім людина помирала.
У район, де спостерігалася куру, вирушила експедиція, яку очолив лікар, антрополог і етнограф Даніел Карлтон Гайду зек, тоді працював в Мельбурні. Експедиція встановила, що хвороба вражає лише тих форе, які жили в джунглях, і не зустрічається у тих дітей цього племені, які жили в місіонерській школі-інтернаті. Члени експедиції ретельно проаналізували більше 400 чинників навколишнього середовища, але не змогли знайти будь-якої отрута або виділити з тканин померлих від куру будь-якого збудника. Єдино, що вдалося встановити, - частіше куру вражає дітей і жінок. Нарешті, один з дослідників настільки подружився з аборигенами, що вони дозволили йому брати участь в ритуалі похорону померлого члена племені. І ось тут-то і з'ясувалося, що небіжчика за місцевим звичаєм з'їдають для того, щоб його розум (мозок), хоробрість (печінка) і сила (м'язи) перейшли до родичів. Причому більшу частину мозку, де, мабуть, знаходився головне джерело інфекції, з'їдали жінки і діти в процесі підготовки «поминок». І хоча інфікується початок визначити, а тим більше виділити, не вдалося, вчені змогли переконати форе відмовитися від такого похоронного ритуалу. Вважається, що аборигени перестали поїдати небіжчиків в 1959 р, але останні випадки куру реєструвалися ще в 1970-ті рр., Що доводить абсолютно неправдоподібну тривалість інкубаційного періоду (прихованого періоду розвитку збудника в організмі).
Подальші спроби зрозуміти механізм виникнення куру, вжиті Гайду- зеком і його співробітниками в кінці 1960-х - початку 1970-х рр., Зустрілися з великими труднощами. Збудника не тільки не вдавалося виділити в чистому вигляді і культивувати в лабораторних умовах, а й просто прищепити його лабораторним тваринам (щурам, морським свинкам), а також вівцям і шимпанзе. І в цьому немає нічого дивного, оскільки час появи перших симптомів, іноді малопомітних, становило від півтора до трьох років.
Коли було доведено, що куру - заразна хвороба, то її віднесли до так званим повільним вірусних інфекцій, так само як і інше ній- родегенератівное захворювання, при якому немає ні запалення, ні лихоманки, - хвороба Кройцфельдта - Якоба. Це захворювання зустрічається по всьому світу у літніх людей з частотою один випадок на мільйон жителів і характеризується деструкцією сірої речовини СМ і ГМ. Воно починається зазвичай в старості і проявляється в наростаючій м'язової ригідності, порушення довільних рухів, мови і ковтання, втрати пам'яті, слабоумство та інших психічних розладах. Було показано, що екстракт такого мозку містить щось, здатне заразити людиноподібних мавп. У 1968 р інфекційний агент хвороби був виділений у шимпанзе, і пізніше хвороба була щеплена кішці і хом'яка. Часовий інтервал між зараженням експериментального тваринного і появою симптомів зазвичай перевищував один рік.
Приблизно в цей же час було встановлено, що схожі за клінічною картиною і даними посмертного розтину хвороби виявлені у овець (хвороба скрейпі), і інкубаційний період також триває не менше року. Екстракт з мозку хворих людей і тварин піддавали найрізноманітнішим впливам при спробах вбити невідомий збудник. Його нагрівали, опромінювали згубним для всього живого ультрафіолетом, обробляли лугами - все Его не знезаражується екстракт, і він продовжував викликати захворювання. Тоді стало ясно, що людство зіткнулося з якимось типом «неживого» збудника, абсолютно не відомим науці.
математик Дж. С. Гріффіт запропонував сміливу гіпотезу: агент взагалі не містить ніякого генетичного матеріалу, він - лише змінена форма клітинного білка. Відтворення властивостей відбувається шляхом аутокаталіз: частинки білка, що викликають захворювання, самі відтворюють себе. Природно, що лікарі і біологи зустріли таке припущення з недовірою. Адже всі відомі досі збудники були клітинними (бактерії, найпростіші гриби) або неклітинні (віруси) формами життя, що містять речовину спадковості - нуклеїнові кислоти. За виділення невловимого збудника взявся американський вірусолог і біохімік Стенлі Бен Прузінер. Він встановив, що скрейпі хворіють хом'яки, у яких інкубаційний період набагато коротше. З мозку хом'яків Прузінер з співробітниками через 10 років важкої роботи виділили білок, який назвали пріоном (Англ, prion, від proteinceons infections particle - білкова інфекційна частка).
Виявилося, що нормальний пріонних білок дуже часто зустрічається на поверхні нейронів, мабуть, приймаючи якусь участь в міжклітинних зв'язках, зокрема в синаптичних контактах, і містить 255 амінокислот. Нормальні молекули білка-приона у здорової людини мають цілком певну конформацію (просторову структуру), в якій переважають а-спіралі. Такий «здоровий» білок позначається РГР і відрізняється нормальної розчинність. Однак з незрозумілих причин в організмі здорової людини або тварини з'являються мутантні поодинокі молекули такого ж але первинну структуру білка, але мають іншу конформацію, в якій менше спіральних ділянок. Такий «інфекційний» білок-пріон позначається РГР30 (Рис. 11.2).
Цей структурний ізомер пріонів білка дуже стійкий і здатний до швидкої полімеризації, т. Е. Утворення угруповань з одиночних молекул, в результаті чого утворюються великі білкові частки. В даний час описано декілька мутацій гена пріонів, які призводять до розвитку нейродегенеративних захворювань.
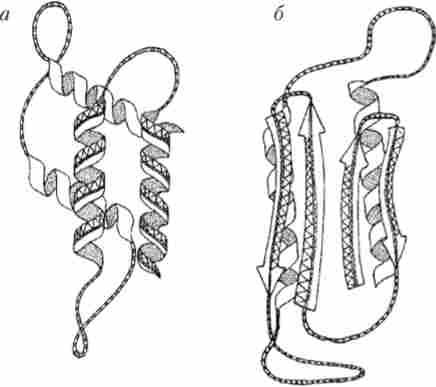
Мал. 11.2. Різниця в конформації пріонів білка при його перетворенні з форми РГРз (А) в патогенну форму PrPsc (Б)
Губчасті енцефалопатії починаються з того, що в організмі з'являється білок-пріон в конформації РГР50. Це може статися в результаті мутації в самій клітині, а може бути результатом індукування, т. Е. Попадання аномальної молекули ззовні. молекули РГР80 взаємодіють з молекулами РГРз і утворюють комплекс, в якому РГР ° змінює свою конформацію і перетворюється в PrPsc. Молекули розходяться і знову утворюють «пари» з РГРз. Процес йде лавиноподібно і, незважаючи на те, що він може початися всього з однієї молекули, поступово вони накопичуються і, взаємодіючи між собою, утворюють великі високомолекулярні комплекси (амілоідонодобние бляшки) (рис. 11.3).
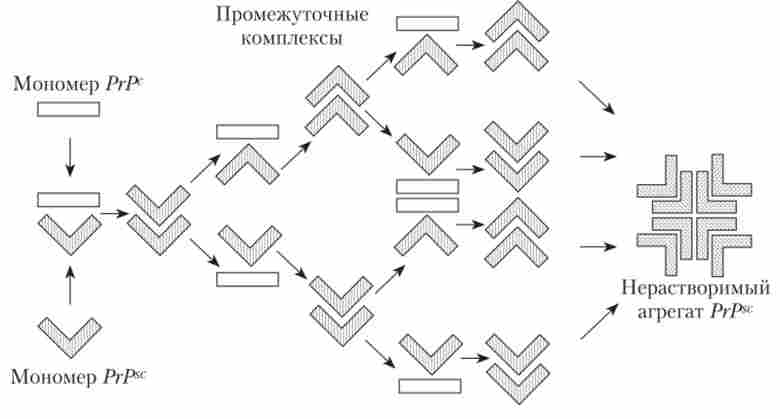
Мал. 113. Схема освіти пріонів білка PrPsc і нерозчинних агрегатів
За кілька років накопичується така кількість бляшок, що вони починають істотно впливати на роботу мозку.
Хвороба Кройцфельдта - Якоба починається з порушення уваги і пам'яті. Людина худне, знижується статевий потяг. Найчастіше перші симптоми спостерігаються у чоловіків у віці 50-65 років. Потім погіршуються зір, координація рухів, прогресує слабоумство. У електроенцефалограмі помітні типові для цього захворювання зміни, за якими фахівці можуть поставити діагноз. Крім того, атрофія мозку помітна на томограмі. Показано, що бляшки сприяють тривалої активації ЛГМДЛ-глутаматних рецепторів до цитотоксичного рівня. Крім того, показано, що РГР * знижує активність ферментів обміну найважливішого нейромедіатора АХ - холінацетилтрансферази і аце- тілхолінестерази. Масова загибель нейронів поступово призводить до повної дезорганізації роботи мозку і загибелі хворого.
Найчастіше в даний час зустрічається спорадична, т. Е. Інфекційна форма хвороби (у 90% хворих).
Ще дві «пріонні» хвороби виникають у людей спонтанно, в результаті мутацій і, мабуть, не є інфекційними. Це синдром Герстманна - Шраусслера - Шейнкера (Гінн) і смертельна сімейна безсоння. Ці хвороби зустрічаються дуже рідко і, будучи спадковими, спостерігаються зазвичай у представників одних і тих же сімей. Наприклад, налічується всього 50 сімей на Землі, члени яких мають мутації, що тягнуть за собою синдром ГШШ. При цьому нейродегенеративних захворюванні особливо багато бляшок вражає мозочок, а тривалість хвороби від появи перших симптомів до смерті становить від двох до шести років.
При смертельної сімейної безсонні в наявності швидкий розвиток губчастого стану мозку, а амілоїдоподібних бляшки практично не розвиваються. Поразки таламуса призводять до порушень сну. Хвороба триває не більше року і закінчується смертю.
Специфічного лікування губчастих енцефалопатій немає. Але при спілкуванні з хворими, мабуть, треба бути обережним, так як шляху передачі пріонів з'ясовані ще недостатньо. Крім того, небезпеку становлять матеріали, взяті для пересадки у хворих людей (рогівка, тверда мозкова оболонка). Необхідний постійний контроль за худобою, оскільки вживати в їжу м'ясо хворих овець і корів смертельно небезпечно.
У 1990-і рр. для прискорення росту коровам в їжу додавали порошок, зроблений головним чином з кісток овець. Потім виявилося, що вівці були хворі скрейпі. У 1992 р в Європі, головним чином в Англії, захворіло губчастої енцефалопатією близько 37 000 тварин. М'ясо багатьох з них було продано в континентальну Європу до того, як з'ясувалося, що корови хворі. Всього в Європі загинуло кілька десятків людей, і економічний збиток склав багато мільйонів доларів (забій худоби, ембарго на торгівлю м'ясом). Росію ця епідемія, мабуть, майже не торкнулася.
Зараз ведуться розробки по створенню специфічних способів захисту від пріонних хвороб. Наприклад, робляться спроби створення способів зворотного перетворення РГР30 в РГРз.
В даному підручнику, природно, неможливо описати всі види і різновиди хвороб, в основі яких лежать розлади роботи ЦНС.
Але наведені вище відомості показують, наскільки важливо навчитися боротися з цими недугами. На жаль, «оживити» нейрони, загиблі в процесі розвитку паркінсонізму або хвороби Альцгеймера - неможливо. Однак робляться спроби «підсадки» в мозок стовбурових клітин, які повинні диференціюватися в нейрони. Відомості про результати цих клінічних експериментів суперечливі, але перші, причому дуже важливі, кроки в цьому напрямку вже зроблені. І якщо відновити деградованих мозок поки неможливо, цілком реально загальмувати розвиток тієї чи іншої хвороби. На цьому шляху досягнуті вражаючі успіхи. Розроблено ліки, що уповільнюють розвиток паркінсонізму. Отримано кошти, що перешкоджають виникненню нападів епілепсії або епізодів депресії. Складні лікарські комплекси дають можливість хворим на шизофренію жити серед здорових людей, «не випадаючи з товариства». Немає сумнівів в тому, що в міру подальших досліджень роботи мозку будуть отримані відомості, які дозволять успішно боротися з нервовими і психічними захворюваннями.